(中国器审 2024-06-09)
一、前言
半月板位于胫骨平台和股骨髁之间,具有传递负荷、吸收震荡、稳定关节等功能,是膝关节的重要结构之一 [1]。半月板损伤在膝关节运动损伤中十分常见,多见于一些激烈的对抗性运动项目中,另外自然老化也会引起半月板整体的破坏[2]。临床上在治疗半月板损伤时通常会根据损伤部位的损伤程度选择合适的修复方式,包括半月板部分或完全切除、半月板缝合、半月板移植等。针对半月板血供丰富区的损伤,临床上通常采用由内而外、由外而内以及全内缝合的半月板缝合术。关节镜下的全内缝合方法,由于其低侵入微创性的优势,成为一种被广泛认可和接受的半月板修复技术。
目前国外很多医疗器械公司均研发了全内半月板缝合系统,在中国也有多年临床应用历史,并取得了良好的疗效[3,4,5]。各个产品系列的操作方法略有区别,但工作原理基本一致。国产全内半月板缝合系统也在产业化进程中,主要以仿制国外进口产品为主。
全内半月板缝合系统通常由植入物和配套工具组成。植入物包括固定锚(棒)和缝线,配套工具包括插入器等,如图1所示。固定锚(棒)通常采用聚醚醚酮材料制成,缝线通常采用超高分子量聚乙烯(或为主)材料制成,插入器通常由手柄和不锈钢材料的针杆制成。
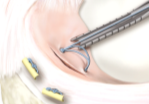
图1.全内半月板缝合系统结构示意图
Fig.1. Schematic diagram of All-inside Meniscus Suture System
二、审评思考
近些年,随着诊疗技术的进步及人们对运动损伤的重视,运动医学植入器械发展迅速,全内半月板缝合系统产业化进程加快,注册申报量大幅增加。但目前全内半月板缝合系统无相关指导原则、标准可参考,一定程度上减缓了产业化进程。通过检索国内外公开资料,结合既往该类产品的技术审评经验,梳理了该类产品比较容易出现问题的审评关注点,主要包括产品基本信息、非临床研究、临床评价、产品技术要求,尤其是非临床研究资料,是注册申报的关注重点。
(一)产品基本信息
在该部分注册申报资料中,申请人需详述全内半月板缝合系统的基本信息和操作原理,通常需包括:(1)正确划分注册单元,作为一个系统配合使用的组件可作为同一注册单元进行申报。(2)阐述产品结构及组成并提供结构示意图,需全面详细地提供全内半月板缝合系统的结构组成信息,包括固定锚(棒)及缝线在配套工具中的部署、固定锚(棒)材质、固定锚(棒)的关键尺寸(如长度、宽度、厚度、直径、过线孔尺寸等)、缝线材质及颜色(包括染料信息)、缝线是否含涂层(涂层材质)、配套工具接触人体部分的材质、灭菌方式及有效期等信息。(3)提供型号规格划分说明,明确不同型号规格之间的差异,可以从固定锚(棒)规格、缝线规格、缝线颜色、关键尺寸(如插入器头部弯曲角度)等方面考虑。(4)明确产品的适用范围、预期适用人群、禁忌证等信息,建议结合产品的预期用途及临床评价资料确定产品的适用范围。(5)提供产品的最小包装单元,包括组件和数量。(6)提供产品的设计依据和作用机理,如明确固定锚(棒)和缝线在配套工具中的部署情况及击发机理,明确在半月板缝合中使用的数量。(7)提供与同类产品的对比信息,建议从适用范围、作用机理、结构及关键尺寸、性能、材料、工艺等方面进行对比,鉴于全内半月板缝合系统包含多个组件,需针对每种组件分别进行结构及关键尺寸对比,对于差异部分需结合临床预期提供其设计依据,必要时通过性能研究论证差异部分不会影响产品的安全有效性。
(二)非临床研究
对于拟上市的全内半月板缝合系统,需提供产品的非临床研究资料,包括功能性、安全性指标以及与质量控制相关的其他指标的确定依据。全内半月板缝合系统的非临床研究需着重从性能研究、生物相容性、动物试验、灭菌确认和稳定性等方面考虑。非临床研究部分也是注册申报的难点,申报资料存在问题较多,申请人需重点关注。
1.性能研究
全内半月板缝合系统的结构设计若不合理将导致产品在临床使用中出现功能失效(如产品发生永久变形导致失效或不能充分承受载荷),无法完成其临床预期功能。开展充分的性能研究,是评估产品功能失效风险的重要手段,也是产品设计验证的重要环节。在全内半月板缝合系统的研发阶段,需根据产品特点及同类产品临床上的不良事件确定相关失效风险源。目前很多国家已建立了医疗器械不良事件监测系统,美国的MAUDE数据库属于全数据库,收录了按照FDA法规进行报告的所有不良事件,信息量较大。通过检索MAUDE数据库,以在境内外同时上市、国产化程度较高的部分半月板缝合系统为目标,汇总了从2016年到2021年的不良事件共520条,针对查询结果逐条进行分析,汇总结果如图2所示。
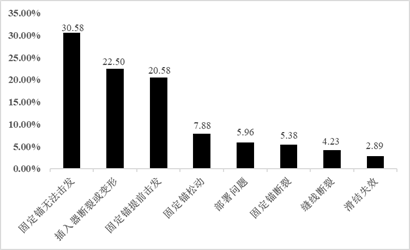
图2. MAUDE数据库全内半月板缝合系统不良事件数据汇总
Fig.2. Summary of adverse event data with All-inside Meniscus Suture System in MAUDE database
根据全内半月板缝合系统不良事件汇总结果,此类产品功能失效的风险点主要集中在固定锚(棒)无法击发或提前击发、插入器断裂或变形、缝线断裂、固定锚(棒)断裂、固定失效等方面,与产品的操作使用性能、插入器强度、缝线强度、系统固定强度直接相关,上述性能也是注册技术审评重点关注的一些指标,下面给出上述性能指标的一些审评思考。
(1)全内半月板缝合系统的操作使用性能:根据不良事件汇总结果,全内半月板缝合系统的主要失效模式是无法正常击发(包括无法击发或提前击发),表明全内半月板缝合术的成功与否很大程度取决于配套手术器械与植入物作为整体的操作使用性能,手术器械对于手术成功与否及手术时间长短有很大影响,手术器械是否操作简便并能正确发挥其预期性能十分关键。针对全内半月板缝合系统进行操作使用测试是完成产品设计确认的工作之一。操作使用性能研究的观察指标通常包括植入物挡板在手术器械中的部署是否合理及能否顺利击发、插入器能否顺利刺穿半月板并不发生断裂或变形、深度限定器能否准确限位、手术器械能否顺利回退、推结器能否顺利完成打结等。在开展操作使用性能研究时可选取离体生物半月板(如猪半月板等)进行模拟临床手术操作,需注意样本量计算、操作人员培训、评价指标设定等内容。
(2)插入器强度:根据不良事件汇总结果,如果插入器断裂或发生较大程度的变形将无法正常击发固定锚(棒)并导致手术的失败,插入器强度是否足够直接决定了缝合系统能否顺利穿透半月板并顺利击发。插入器强度可通过刚性、韧性、刺穿力指标进行量化。目前国内外尚未建立全内半月板缝合系统相关标准,在开展该部分研究时可参考具有类似功能的手术器械标准中的方法,如YY/T 0043-2016《医用缝合针》[6]、YY/T 1148-2009《腰椎穿刺针》[7]等。
(3)缝线强度:根据不良事件汇总结果,全内半月板缝合系统在收紧两个固定锚(棒)时会有缝线断裂的风险,从而导致固定失效。因此,需保证缝线具有足够的强度。缝线强度可通过缝线的断裂强力指标进行量化,其测试方法可参考YY/T 1832-2022《运动医学植入器械 缝线拉伸试验方法》[8]。
(4)系统固定强度:根据不良事件汇总结果,系统固定失效的表现形式多样,包括固定锚(棒)从半月板中脱出、固定锚(棒)断裂、缝线断裂、滑结失效、缝线与固定锚(棒)分离等等,上述失效均可导致半月板缝合手术失败。因此,系统固定强度主要考虑全内半月板缝合系统在手术即刻及愈合过程中的足够稳定性。系统固定强度可通过疲劳前后固定强度、疲劳后缝合处的位移量进行量化。建议模拟临床使用及产品预期作用期限制定相应的试验加载载荷和循环周期等参数,部分文献也给出了一些试验方法(如试验模型、加载载荷、频率和加载周期)可供参考 [9,10]。考虑到体外测试受到诸多试验条件的限制,如半月板模型的选择、加载模型及加载方式、测试环境等,体外测试无法完全模拟体内的情况。针对该性能进行体外测试主要是完成产品设计验证的工作。可以对比不同生产商相似设计的产品的性能,力学测试结果的可接受性建议优先选择结构、尺寸及材料相近的同类已上市产品进行对比,此外也可以接受有学术共识或文献支持性依据的结果。无论选择哪种方法,需注意试验方法的一致性,如半月板模型选取、加载载荷和加载周期,确保试验结果具有可比性。
2.生物相容性
全内半月板缝合系统包括植入类组件(如固定锚(棒)和缝线)和与人体短暂接触类组件(如配套工具等)。预期与人体接触的部分,需要根据接触类型开展生物学评价,必要时进行生物学试验,建议根据GB/T 16886.1-2022《医疗器械生物学评价 第1部分: 风险管理过程中的评价与试验》[11]系列标准对产品的生物相容性进行评价。可参考《医疗器械生物学评价和审查指南》[12](国食药监械[2007]345号)的相关要求阐明实施或豁免生物学试验的理由。
该部分内容需要注意的是,在既往的注册申报过程中,经常也会遇到采用原材料的生物学试验报告完成申报产品的生物学评价的情形,如采用纱线的生物学试验报告完成缝线的生物学评价,需要论证从纱线编织成缝线的生产工艺过程及后续包装灭菌过程不会引入新的生物学风险。
3.动物试验
动物试验是评价医疗器械安全有效的重要手段之一,针对全内半月板缝合系统是否开展动物试验建议参考《医疗器械动物试验研究技术审查指导原则第一部分:决策原则》[13]。对于多数的不可吸收全内半月板缝合系统,如果通过理化表征、性能验证等手段证明其技术特性与市售同类产品具有等同性,通常不需要进行动物试验。
4.灭菌确认和稳定性
全内半月板缝合系统一般以无菌状态交付,灭菌方式通常采用辐照灭菌或环氧乙烷灭菌。申请人需提供灭菌确认资料,具体要求可参考GB 18279[14]、GB 18280[15]系列标准。此外,无菌状态交付的产品需按照《无源植入性医疗器械稳定性研究指导原则》[16]提供稳定性研究资料,可采用实时老化或加速老化的方法。需要注意的是,对于多组件的产品可能存在不同包装的情形,这种情况下应针对每种包装分别提交资料。
(三)临床评价
全内半月板缝合系统需按照《医疗器械临床评价技术指导原则》[17]及《免于进行临床评价的医疗器械目录》来选择合适的临床评价路径。对于多数的全内半月板缝合系统而言,可免于进行临床评价。但如果通过非临床研究无法证明产品的临床安全有效性或者在动物试验中发现仍需要提供临床证据的情况下,有可能需要通过开展申报产品的临床试验以完成临床评价。
(四)全内半月板缝合系统产品技术要求
产品技术要求需参考《医疗器械产品技术要求编写指导原则》[18]的相关要求编制。产品技术要求需符合相关标准及法律法规的要求,同时也要结合产品的设计属性制定保证产品安全、有效的产品技术要求,全内半月板缝合系统技术要求中的性能指标可以参考以下列举的项目进行制定,并结合申报产品特点选择或增加适用的性能指标:
1.固定锚(棒):外观、表面粗糙度、表面缺陷、尺寸、硬度(如适用);
2.缝线:外观、线径、长度、断裂强力、褪色试验;
3.插入器:刺穿力、外观、表面粗糙度、硬度、耐腐蚀性能;
4.系统固定强度、无菌、环氧乙烷残留量。
此外,金属材料的化学成分和显微组织、聚合物材料的理化性能、阳极氧化表面元素定性分析等适用内容应在产品技术要求附录中予以规定,并提供相应的支持性资料。
该部分内容需要注意的是,技术要求中性能指标的制定要全面,同时需根据产品临床需求和生产质控水平制定性能指标的可接受限值,比如在既往的审评过程中,申请人在制定缝线的断裂强力指标接受限值时,往往直接采用YY 0167-2020《非吸收性外科缝线》[19]中的接受限值,但全内半月板缝合系统中的缝线与外科缝线的预期用途存在较大差异,其设计预期并非单纯用于人体组织缝合,而需承担固定半月板的拉力,与外科缝线在缝线强度上要求不同,因此,审评过程中会要求申请人结合产品临床需求和生产质控水平重新制定缝线断裂强力的接受限值。
三、结语
本文针对全内半月板缝合系统无相关标准或指导原则可参考的现状,总结了该类产品的技术审评关注点,提出监管思考,供研究者参考,以期提高产品注册申报资料质量和监管效能,加快相关产品的产业化进程。建议申请人提高研发设计能力,开展科学合理的性能研究以保证产品的安全有效,同时也为支持注册申报提供充分的证据。此外,加快制定该类产品的相关标准或指导原则很有必要,有助于进一步积累监管数据,推进医疗器械大数据构建进程,减轻企业负担的同时提高监管的智能化水平,促进产业创新发展。
参考文献:
[1] 耿晓林,周迎峰,张超,等.关节镜下半月板部分切除术治疗膝关节半月板损伤的临床研究[J].创伤外科杂志,2020,22(03):212-216.
[2] 江佩师,陈志伟,方玉基,等.602例膝关节半月板损伤流行病学调查[J].中南医学科学杂志,2020,48(02):160-163.
[3] 王小武,黄晓华,张鹏,等.关节镜下应用FasT-Fix缝合器治疗半月板桶柄样撕裂损伤[J].临床骨科杂志,2019,22(04):437-439.
[4] 李政,王平,李长树等:关节镜下AR缝合枪缝合外侧半月板撕裂的临床疗效观察[J].实用骨科杂志,2020,26(1):18-22.
[5] 刘聚,明立德,许建中,等.关节镜下Omnispan系统与FAST-fix 360°系统缝合修复内外侧半月板损伤的疗效比较[J].中国骨与关节损伤杂志,2019,34(02):165-167.
[6] 国家食品药品监督管理总局,全国外科器械标准化技术委员会. 医用缝合针: YY/T 0043-2016 [S]. 北京:中国标准出版社,2017.
[7] 国家食品药品监督管理局,全国医用注射器(针)标准化技术委员会. 腰椎穿刺针:YY/T 1148-2009[S]. 北京:中国标准出版社,2009.
[8] 国家药品监督管理局,全国外科植入物和矫形器械标准化技术委员会. 运动医学植入器械 缝线拉伸试验方法:YY/T 1832-2022[S]. 北京:中国标准出版社,2022.
[9] Zantop T, Eggers AK, Musahl V, et al. Cyclic testing of flexible all-inside meniscus suture anchors: Biomechanical analysis. Am J Sports Med 2005;33:388.
[10] Jen-Huei Chang, Hsain-Chung Shen, Guo-Shu Huang,et al. A Biomechanical Comparison of All-Inside Meniscus Repair Techniques, Journal of Surgical Research 155, 82–88 (2009).
[11] 国家市场监督管理总局国家标准化管理委员会. 医疗器械生物学评价第1部分:风险管理过程中的评价与试验[S]. 北京:中国标准出版社,2022.
[12] 国家食品药品监督管理局.关于印发医疗器械生物学评价和审查指南的通知 [EB/OL]. (2007-06-15)[2022-10-15]. https://www.nmpa.gov.cn/xxgk/fgwj/gzwj/gzwjylqx/20070615010101796.html.
[13] 国家药品监督管理局. 医疗器械动物试验研究技术审查指导原则第一部分:决策原则(2021年修订版) [EB/OL]. (2020-09-27) [2022-10-15]. https://www.nmpa.gov.cn/xxgk/ggtg/qtggtg/20210927153130147.html.
[14] 中华人民共和国国家质量监督检验检疫总局中国国家标准化管理委员会. 医疗保健产品灭菌 环氧乙烷: GB 18279.1~2-2015 [S]. 北京:中国标准出版社,2016.
[15] 中华人民共和国国家质量监督检验检疫总局中国国家标准化管理委员会. 医疗保健产品灭菌 辐射: GB 18280.1~3-2015 [S]. 北京:中国标准出版社,2016.
[16] 国家药品监督管理局医疗器械技术审评中心.无源植入性医疗器械稳定性研究指导原则(2022年修订版) [EB/OL].(2022-03-16)[2022-10-15]. https://www.cmde.org.cn//xwdt/shpgzgg/gztg/20220316164500666.html.
[17] 国家药品监督管理局.医疗器械临床评价技术指导原则[EB/OL].(2021-09-28)[2022-10-15]. https://www.nmpa.gov.cn/xxgk/ggtg/qtggtg/20210928170338138.html.
[18] 国家药品监督管理局.医疗器械产品技术要求编写指导原则 [EB/OL]. (2022-02-09)[2022-10-15]. https://www.nmpa.gov.cn/xxgk/ggtg/qtggtg/20220209152322130.html.
[19] 国家药品监督管理局,全国外科器械标准化技术委员会. 非吸收性外科缝线: YY 0167-2020 [S]. 北京:中国标准出版社,2020.
器械大湾区分中心 高进涛 供稿
(转载自国家药品监督管理局医疗器械技术审评中心网站)