(中国器审 2024年10月24日)
一、背景介绍
校准品(Calibrator),主要用于校准检测仪器和检测方法,以保证检测结果的准确性和一致性,其作为校准函数中用作独立变量值的参考物质,是体外诊断试剂或检测系统中的重要组成部分,也是实现体外诊断试剂临床检测及监督检验结果准确一致的主要工具,对于整个体外诊断检测系统中量值的传递及结果测试的准确起到至关重要的作用。
校准品与试剂配合使用,常见于定量检测体外诊断试剂,一般包含若干个具有不同的特定值浓度的校准品点,通过校准品的系列浓度与信号之间关系,为检测系统建立或调整拟合校准体系,通过校准曲线将样本测试信号计算转换为最终的浓度结果。
从形态上,校准品可为液体或固体冻干粉状态,储存条件与原材料性质相关,通常为2~8℃冷藏、-20℃冷冻或15~25℃室温保存;从项目上,校准品可分为单个项目校准品或多项目复合校准品,多项复合校准品通常包含多个不相互干扰,且有一定关联的项目产品,如肿瘤标志物复合校准品、酶类复合校准品等。
二、校准品量值溯源
量值溯源也称计量溯源性( metrological traceability),是指通过一条形成文件的、具有规定测量不确定度及不间断的校准链,使测量结果与参考标准联系起来的特性。依据国际标准化组织最新发布的《ISO 17511:2020 体外诊断医疗器械.校准品 真实性控制物质和人体样品值的计量溯源性要求》,体外诊断试剂量值溯源的校准等级主要可以分为六类:
1.具有参考测量程序(Reference Measurement Procedure, RMP)和一级参考物质(Reference Materials, RM),为最理想的量值溯源模式,通常见于定义明确的小分子物质,如电解质类物质(钾、钠、钙离子等)、代谢类物质(葡萄糖、肌酐等)以及部分固醇激素、甲状腺激素等;
2.具有一级 RMP,无一级 RM,包括酶学项目、部分凝血因子等;
3.具有经特定的一级校准品校准的 RMP,如糖化血红蛋白A1c。
4.有国际约定校准品的情况,适用于具有国际约定校准品的被测量,常见有WHO国际标准物质的项目,如肾素、癌胚抗原等;
5.具有国际一致化方案支持,可适用于存在有证参考物质或国际约定校准品,但由于缺乏互换性而导致不适用的情况,如蛋白激素类、部分肿瘤标志物等,具体可参照 ISO 21151的要求;
6.被测量只溯源到制造商内部人为规定的RM,即上述五种情况以外的待测物,如部分肿瘤标志物及大部分抗体类等。
前三种校准情形可通过一级参考测量程序(RMP)和(或)一个或多个有证参考物质(RM)的支持,测量结果可溯源至国际单位(SI),后三种无法通过参考测量程序(RMP)或有证参考物质(RM)支持,测量结果不可溯源至SI。
在这六种不同的校准等级中,无论溯源至SI、RMP或RM的任何情况,在体外诊断试剂制造商及终端用户的量值传递中都会存在校准品的参与。在不同的校准等级中存在不同的校准品,工作校准品的功能通常是使用多个浓度(多为6~10个)的校准品建立校准体系主曲线,用于检测试剂的曲线拟合以及样本结果定量计算,产品校准品的功能通常是使用若干个(2~6个)校准品对已经在制造商内部建立的主曲线进行调整优化,降低或消除测试系统由于不同时间、地点以及测试环境带来的干扰或影响。通常这种校准计算方式比较常见,不过也有企业不直接建立内置主曲线,直接在终端客户测试阶段采用产品校准品来绘制校准曲线进行浓度计算。
在产品的量值溯源过程中,IVD制造商选定的参考测量程序经过上一级的参考物质的校准,将量值及其不确定度传递至工作校准品或主校准品,然后再通过制造商常规测量程序,为终端用户校准品或产品校准品赋值,最后通过校准终端用户用仪器,将量值传递至样本结果。
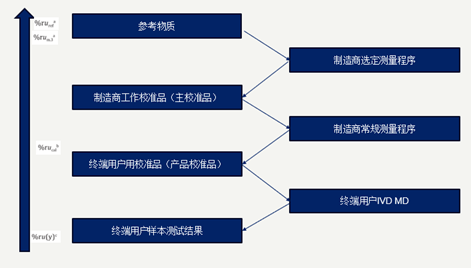
图1.量值溯源中的校准等级
三、校准品注册上市情况
校准品可以与配合使用的体外诊断试剂合并申请注册,也可以单独申请注册。按照《体外诊断试剂注册与备案管理办法(国家市场监督管理总局令第48号)》的要求,校准品应依据其预期用途进行命名,其产品名称通常为XX校准品,与配套使用的试剂盒及预期待测物质保持一致。
从产品的预期使用风险角度来看,二、三类体外诊断试剂校准品的管理类别通常与配合使用的试剂管理类别一致,如校准品可配合多个不同管理类别的试剂盒产品使用时,其管理类别与试剂盒中管理类别最高级的产品保持一致。
通过检索已经批准上市的产品,已批准的试剂产品超过15000个,而单独注册的校准品产品仅为700+,大部分企业将校准品与试剂盒合并申报。
四、校准品注册审评关注要点
校准品通常非单独使用,需要配套试剂进行检测,其性能指标大部分都是通过试剂的性能来体现,此外校准品单独申请注册不需要提交临床评价资料。
校准品性能指标要求相对较少,主要包括稳定性研究、均匀性、精密度等。稳定性研究应包含实时稳定性、使用稳定性和运输稳定性,如校准品为冻干粉末状态,还应提交复溶后的稳定性研究资料。
校准品可以使用血清、血浆、尿液等生物源性基质,应尽可能与实际临床测试样品的基质保持一致,使校准结果更接近实际情况,避免校准品与真实样本之间的基质差异问题。如校准品基质和样品存在差异,还应进行校准品的互换性研究。
校准品在体外诊断中起到非常重要的作用,它们的质量和准确性直接影响着整个诊断过程的可靠性和准确性。目前暂无有关单独校准品的技术审查指导原则,企业在产品注册申报过程中,参考相关指导原则中关于校准品部分的要求。后续可对审评过程中的重点关注问题进行总结规范,为注册审评提供指导参考依据,也为企业的设计开发及注册申报提供指导和规范。
参考文献:
【1】 ISO 17511:2020《In vitro diagnostic medical devices Requirements for establishing metrological traceability of values assigned to calibrators, trueness control materials and human samples》
【2】CNAS-CL01-G002:2021《测量结果的计量溯源性要求》
器械长三角分中心 孙贇 供稿
(转载自国家药品监督管理局医疗器械技术审评中心网站)